Délétion 1q21.1
Les informations contenues dans ce résumé du Syndrome de délétion 1q21.1 proviennent de publications de recherche. Il ne s’agit pas d’un avis médical.
Cliquez ici pour consulter notre guide complet des gènes de la délétion 1q21.1
Le Guide des gènes en ligne comprend des informations supplémentaires sur la délétion 1q21.1, telles que le risque d’avoir un autre enfant atteint de cette maladie, les problèmes de comportement et de développement liés à la délétion 1q21.1 ou les spécialistes à prendre en compte pour les personnes atteintes de cette maladie. Partagez cette ressource avec les membres de votre famille ou vos prestataires de soins.
Le syndrome de la délétion 1q21.1 est également appelé syndrome de microdélétion 1q21.1. Dans cette page web, nous utiliserons le nom “syndrome de délétion 1q21.1”. syndrome de délétion 1q21.1 pour englober le large éventail de variantes observées chez les personnes identifiées.
Le dernier rapport Simons Searchlight contient des informations actualisées sur votre communauté génétique. Il met en évidence la puissance de vos contributions et la manière dont ces données font progresser la recherche.
Si vous n’êtes pas encore inscrit, pensez à rejoindre Simons Searchlight pour figurer dans les prochains rapports !
Consultez tous les rapports ci-dessous en cliquant sur “Rapports trimestriels précédents” au bas de cette page.

Qu’est-ce que le syndrome de délétion 1q21.1?
Le syndrome de délétion 1q21.1 se produit lorsqu’il manque à une personne un morceau du chromosome 1, l’un des 46 chromosomes de l’organisme. Les chromosomes sont des structures de nos cellules qui abritent nos gènes.
Rôle clé
La région de délétion 1q21.1 joue un rôle dans le développement du cerveau.
Symptômes
La région de la délétion 1q21.1 jouant un rôle important dans le développement et le fonctionnement du cerveau, de nombreuses personnes atteintes du syndrome de la délétion 1q21.1 sont atteintes de la maladie d’Alzheimer :
- Faible tonus musculaire
- Retard global de développement
- Handicap intellectuel
- Petite taille de tête
- Faible croissance
- Problèmes de comportement tels que l’agressivité
- Trouble du déficit de l’attention/hyperactivité (TDAH)
- Problèmes de sommeil
- Autisme
- Crises d’épilepsie
- Problèmes de vision, tels que le strabisme (yeux croisés) ou le nystagmus (mouvements répétitifs incontrôlés des yeux).
- Perte auditive
- Laxité articulaire
- Dysmorphie faciale légère mais non spécifique
Combien de personnes sont atteintes du syndrome de délétion 1q21.1?
En 2024, au moins 102 personnes atteintes du syndrome de délétion 1q21.1 ont été identifiées par la recherche médicale. Le premier cas a été découvert en 2008. Il y a probablement beaucoup d’autres personnes non diagnostiquées qui sont atteintes de ce syndrome.
Pour en savoir plus sur la délétion 1q21.1 et entrer en contact avec d’autres familles de Simons Searchlight, consultez les ressources ci-dessous.
Ressources de soutien
- CommunautéSimons Searchlight – Groupe Facebook « Délétion 1q21.1 »
- Gène SFARI – Délétion 1q21.1
Information génétique
Quels sont les gènes impliqués dans les délétions et duplications 1q21.1 typiques (classe 1) ? 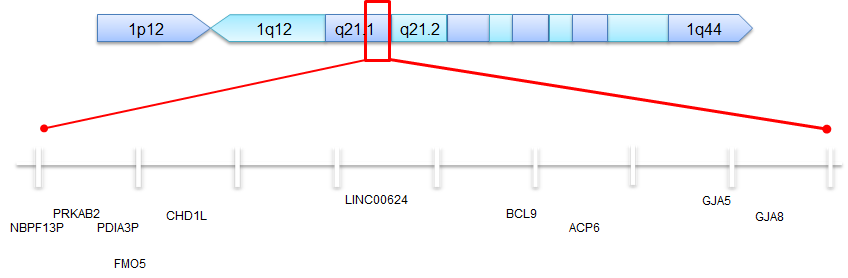 Dans une délétion ou une duplication 1q21.1 “typique”, il y a environ 20 gènes manquants (supprimés) ou supplémentaires (dupliqués). Certaines n’ont fait l’objet d’aucune recherche et ont été omises dans le résumé. Ces gènes commençant par “NBPF” ont été découverts chez des enfants présentant des caractéristiques telles que la macrocéphalie, l’autisme, la schizophrénie, la déficience intellectuelle, la cardiopathie congénitale, le neuroblastome et des problèmes au niveau des reins et des voies urinaires. Cette famille de gènes contient de nombreux
Dans une délétion ou une duplication 1q21.1 “typique”, il y a environ 20 gènes manquants (supprimés) ou supplémentaires (dupliqués). Certaines n’ont fait l’objet d’aucune recherche et ont été omises dans le résumé. Ces gènes commençant par “NBPF” ont été découverts chez des enfants présentant des caractéristiques telles que la macrocéphalie, l’autisme, la schizophrénie, la déficience intellectuelle, la cardiopathie congénitale, le neuroblastome et des problèmes au niveau des reins et des voies urinaires. Cette famille de gènes contient de nombreux
- PRKAB2 – Joue un rôle important dans la régulation de l’énergie au sein des cellules (impliqué dans la synthèse des acides gras et du cholestérol pour les cellules)
- FMO5 – L’absence de deux copies de ce gène est associée à une affection appelée « triméthylaminurie », qui se caractérise par une odeur inhabituelle.
- GJA8 – Ce gène joue un rôle important dans la croissance de certaines cellules de l’œil appelées « cellules fibreuses du cristallin ». L’absence de copies de ce gène (délétions) peut entraîner un risque accru de cataracte.
- CHD1L – Joue un rôle important dans le remodelage de la chromatine (il s’agit en quelque sorte d’un travail « en coulisses » consistant à préparer l’ADN en vue de son utilisation, de sa lecture ou de sa copie).
- BCL9 – La fonction de ce gène reste inconnue lorsqu’il est délété ou dupliqué chez un individu. Des anomalies de ce gène ont été mises en évidence chez des patients atteints de leucémie lymphoblastique aiguë à cellules B. On pense que ces anomalies génétiques surviennent dans les cellules cancéreuses d’un individu (qui présentent généralement des caractéristiques génétiques différentes de celles des cellules du reste de l’organisme). Nous ne pensons pas que les personnes présentant des délétions ou des duplications de ce gène courent un risque accru de leucémie.
Qu’est-ce qu’un pseudogène ? Les pseudogènes sont considérés comme des gènes inactifs. (“Pseudo” signifie “faux” en latin). Ils sont similaires aux gènes normaux, mais ne sont pas fonctionnels. L’information ou “séquence génétique” d’un pseudogène est souvent similaire à un autre gène qui a une fonction dans l’organisme. Les pseudogènes apparaissent à la suite d’erreurs de copie qui se sont produites dans le passé. Il s’agit en fait de copies d’autres gènes fonctionnels, qui ont été incorporés dans l’ADN d’une personne il y a très longtemps, mais qui n’ont plus d’utilité. Les pseudogènes sont considérés comme de l’ADN “non codant”.
GeneReviews
GeneReviews est une excellente ressource à présenter aux cliniciens de votre enfant. Ces publications fournissent un résumé des recherches actuelles sur les maladies génétiques et des informations sur les soins en cours.
- Chapitre de GeneReviews pour 1q21.1 Deletions
Résumés d’articles de recherche
Nous avons résumé ci-dessous des articles de recherche consacrés à la délétion 1q21.1. Nous espérons que ces informations vous seront utiles. Les données disponibles concernant la délétion 1q21.1 sont limitées, et les familles ainsi que les médecins ont un besoin crucial d’en savoir davantage. À mesure que nous en apprendrons davantage grâce aux enfants présentant une mutation de ce gène, nous espérons que cette liste de ressources et d’informations s’étoffera.
Les versions intégrales des articles de recherche publiés sont disponibles sur PubMed. PubMed est une base de données en ligne gratuite gérée par les Instituts nationaux de la santé (NIH). Elle regroupe des articles de recherche tant médicaux que scientifiques. Vous pouvez effectuer une recherche sur PubMed concernant les articles relatifs à la délétion 1q21.1 en cliquant ici.
- Phénotype clinique de la variante récurrente du nombre de copies 1q21.1 Article de recherche original de Bernier R. et al. (2016). Lisez l’article ici et le résumé de Simons Searchlight ici. Lire la couverture médiatique de l’article ici.
Possibilités de recherche
Simons Searchlight Aidez l’équipe de Simons Searchlight à mieux comprendre la délétion 1q21.1 en participant à notre recherche. Vous pouvez en savoir plus sur le projet et vous inscrire ici.
Opportunité de recherche externe : FaceMatch
FaceMatch est une plateforme qui aide les parents et les médecins à contribuer à une base de données internationale sécurisée d’images d’enfants diagnostiqués et non diagnostiqués dans le monde entier. *Cette étude n’est pas affiliée à Simons Searchlight. En savoir plus sur FaceMatch.
Histoires de famille
Rapports trimestriels précédents
- Rapport sur la voix de la communauté 2021
- Rapport trimestriel 2021 sur la suppression du gène 1q21.1
- 1q21.1 Deletion Rapport trimestriel 2 2021
- Suppression du gène 1q21.1 Rapport du troisième trimestre 2021
- 1q21.1 Deletion Quarter 4 Report 2021
- 1q21.1 Deletion Rapport trimestriel 2022
- 1q21.1 Deletion Rapport trimestriel 2 2022
- Suppression du gène 1q21.1 Rapport du troisième trimestre 2022
- 1q21.1 Suppression Rapport du 4e trimestre 2022/trimestre 1 2023
- Rapport sur la suppression du gène 1q21.1 au cours du deuxième trimestre 2023
- Rapport sur la suppression du gène 1q21.1 au troisième trimestre 2023
- 1q21.1 Suppression Trimestre 4 2023/Trimestre 1 2024 Rapport
- Rapport sur la suppression du gène 1q21.1 au cours du deuxième trimestre 2024
- Rapport sur la suppression du gène 1q21.1 au troisième trimestre 2024
- 1q21.1 Deletion Quarter 4 2024 Report – 2024 Surveys Spotlight
- 1q21.1 Deletion Quarter 1 2025 Report – Vineland Spotlight
- Rapport sur la délétion du gène 1q21.1 au cours du deuxième trimestre 2025
- Rapport sur la suppression du gène 1q21.1 au cours du troisième trimestre 2025
- Délétion 1q21.1 : Rapport du 4e trimestre 2025 / 1er trimestre 2026