1q21.1 Deletion
Die Informationen für diese Zusammenfassung von 1q21.1-Deletionssyndrom stammt aus Forschungsveröffentlichungen. Dies ist nicht als Ersatz für eine ärztliche Beratung gedacht.
Klicken Sie hier für unseren vollständigen 1q21.1-Deletionsgenführer
Der Online-Genführer enthält weitere Informationen über die 1q21.1-Deletion, z. B. über die Wahrscheinlichkeit, ein weiteres Kind mit dieser Erkrankung zu bekommen, über Verhaltens- und Entwicklungsprobleme im Zusammenhang mit der 1q21.1-Deletion oder über Spezialisten, die für Menschen mit dieser Erkrankung in Frage kommen. Teilen Sie diese Ressource mit Familienmitgliedern oder Ihren medizinischen Betreuern.
1q21.1-Deletionssyndrom
wird auch als 1q21.1 Mikrodeletionssyndrom. Für diese Webseite verwenden wir den Namen
1q21.1-Deletionssyndrom
um das breite Spektrum der bei den identifizierten Personen beobachteten Varianten zu beschreiben.
Der neueste Simons Searchlight-Bericht enthält aktuelle Informationen über Ihre genetische Gemeinschaft. Es zeigt, wie wichtig Ihre Beiträge sind und wie diese Daten die Forschung voranbringen.
Wenn Sie noch nicht registriert sind, sollten Sie sich bei Simons Searchlight anmelden, um in künftigen Berichten berücksichtigt zu werden!
Sehen Sie sich alle Berichte unten an, indem Sie auf “Frühere Quartalsberichte” am Ende dieser Seite klicken.

Was ist das 1q21.1-Deletionssyndrom?
Das 1q21.1-Deletionssyndrom tritt auf, wenn jemandem ein Stück des Chromosoms 1 fehlt, eines der 46 Chromosomen des Körpers. Chromosomen sind Strukturen in unseren Zellen, die unsere Gene beherbergen.
Schlüsselrolle
Die Deletionsregion 1q21.1 spielt eine Rolle bei der Gehirnentwicklung.
Symptome
Da die 1q21.1-Deletionsregion für die Entwicklung und Funktion des Gehirns wichtig ist, haben viele Menschen mit dem 1q21.1-Deletionssyndrom:
- Niedriger Muskeltonus
- Globale Entwicklungsverzögerung
- Geistige Behinderung
- Kleine Kopfgröße
- Schlechtes Wachstum
- Verhaltensauffälligkeiten wie Aggression
- Aufmerksamkeitsdefizit-/Hyperaktivitätsstörung (ADHS)
- Schlafprobleme
- Autismus
- Krampfanfälle
- Sehstörungen wie Strabismus (Schielen) oder Nystagmus (wiederholte unkontrollierte Augenbewegungen)
- Gehörverlust
- Laxheit der Gelenke
- Milde, aber unspezifische dysmorphe Gesichtszüge
Wie viele Menschen haben das 1q21.1-Deletionssyndrom?
Bis zum Jahr 2024 wurden in der medizinischen Forschung mindestens 102 Menschen mit dem 1q21.1-Deletionssyndrom identifiziert. Der erste Fall wurde im Jahr 2008 entdeckt. Wahrscheinlich gibt es noch viel mehr Menschen, bei denen das Syndrom nicht diagnostiziert wurde.
Erfahren Sie mehr über die 1q21.1-Deletion und schließen Sie sich mit anderen Simons-Searchlight-Familien an (siehe unten).
Ressourcen unterstützen
- Simons Searchlight Community – Facebook-Gruppe „1q21.1-Deletion“
- SFARI-Gen – 1q21.1-Deletion
Genetische Informationen
Welche Gene sind an typischen (Klasse 1) 1q21.1-Deletionen und -Duplikationen beteiligt? 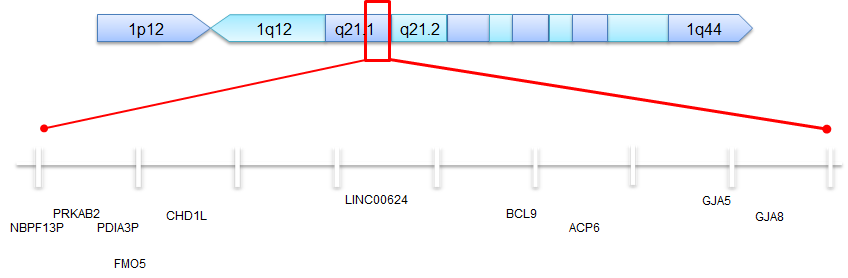 Bei der “typischen” 1q21.1-Deletion oder -Duplikation fehlen etwa 20 Gene (gelöscht) oder sind zusätzlich (dupliziert) vorhanden. Einige wurden überhaupt nicht recherchiert und sind in der Zusammenfassung nicht berücksichtigt worden. Diese Gene, die mit “NBPF” beginnen, wurden bei Kindern mit Merkmalen wie Makrozephalie, Autismus, Schizophrenie, geistiger Behinderung, angeborenem Herzfehler, Neuroblastom und Problemen mit den Nieren und Harnwegen gefunden. Diese Genfamilie enthält viele Pseudogene (siehe Definition unten).
Bei der “typischen” 1q21.1-Deletion oder -Duplikation fehlen etwa 20 Gene (gelöscht) oder sind zusätzlich (dupliziert) vorhanden. Einige wurden überhaupt nicht recherchiert und sind in der Zusammenfassung nicht berücksichtigt worden. Diese Gene, die mit “NBPF” beginnen, wurden bei Kindern mit Merkmalen wie Makrozephalie, Autismus, Schizophrenie, geistiger Behinderung, angeborenem Herzfehler, Neuroblastom und Problemen mit den Nieren und Harnwegen gefunden. Diese Genfamilie enthält viele Pseudogene (siehe Definition unten).
- PRKAB2 – Wichtig für die Energieregulierung in Zellen (beteiligt an der Bildung von Fettsäuren und Cholesterin für die Zellen)
- FMO5 – Das Fehlen von zwei Kopien dieses Gens steht im Zusammenhang mit einer Erkrankung namens „Trimethylaminurie“, die einen ungewöhnlichen Geruch verursacht.
- GJA8 – Spielt eine wichtige Rolle für das Wachstum bestimmter Zellen im Auge, die als „Linsenfasenzellen“ bezeichnet werden. Fehlen Kopien dieses Gens (Deletionen), kann dies zu einem erhöhten Risiko für den Grauen Star führen.
- CHD1L – Spielt eine wichtige Rolle beim Chromatin-Umbau (dies entspricht sozusagen der „Arbeit hinter den Kulissen“, bei der die DNA für die Nutzung, das Ablesen oder die Vervielfältigung vorbereitet wird).
- BCL9 – Die Funktion dieses Gens ist unbekannt, wenn es bei einer Person deletiert oder dupliziert ist. Bei Patienten mit akuter B-Zell-Lymphoblastenleukämie wurden Fehlbildungen in diesem Gen festgestellt. Es wird angenommen, dass die Mutationen in diesem Gen in den Krebszellen einer Person auftreten (die sich in der Regel genetisch von den Zellen im übrigen Körper unterscheiden). Wir gehen nicht davon aus, dass Menschen mit Deletionen oder Duplikationen dieses Gens ein erhöhtes Leukämierisiko aufweisen.
Was ist ein Pseudogen? Pseudogene gelten als inaktive Gene. (“Pseudo” bedeutet auf Lateinisch “Fälschung”.) Sie ähneln normalen Genen, sind aber nicht funktionsfähig. Die Information oder “genetische Sequenz” eines Pseudogens ähnelt oft einem anderen Gen, das eine Aufgabe im Körper hat. Pseudogene entstehen aufgrund von Kopierfehlern, die in der Vergangenheit passiert sind. Sie sind eigentlich Kopien anderer funktionierender Gene, die vor sehr langer Zeit in die DNA eines Menschen eingebaut wurden, aber keine Aufgabe mehr haben. Pseudogene werden als “nicht codierende” DNA betrachtet.
GeneReviews
GeneReviews sind eine hervorragende Ressource, die Sie den Ärzten Ihres Kindes vorlegen können. Diese Veröffentlichungen bieten eine Zusammenfassung der aktuellen Forschung über genetische Erkrankungen und Informationen über die laufende Betreuung.
- GeneReviews Kapitel für 1q21.1 Deletionen
Zusammenfassungen von Forschungsartikeln
Im Folgenden haben wir Forschungsartikel zum Thema 1q21.1-Deletion zusammengefasst. Wir hoffen, dass diese Informationen für Sie hilfreich sind. Die verfügbaren Informationen zur 1q21.1-Deletion sind begrenzt, und sowohl Familien als auch Ärzte haben einen dringenden Bedarf an weiteren Erkenntnissen. Da wir durch die Beobachtung von Kindern mit einer Veränderung in diesem Gen immer mehr erfahren, gehen wir davon aus, dass diese Liste mit Ressourcen und Informationen weiter wachsen wird.
Die Volltexte veröffentlichter Forschungsartikel finden Sie auf PubMed. PubMed ist eine kostenlose Online-Datenbank der National Institutes of Health (NIH). Sie umfasst sowohl medizinische als auch naturwissenschaftliche Forschungsartikel. Eine PubMed-Suche nach Artikeln zur 1q21.1-Deletion finden Sie hier.
- Klinischer Phänotyp der rezidivierenden Kopienzahlvariante 1q21.1 Original-Forschungsartikel von Bernier R. et al. (2016). Lesen Sie den Artikel hier und die Zusammenfassung von Simons Searchlight hier. Lesen Sie die Berichterstattung über diesen Artikel hier.
Forschungsmöglichkeiten
Simons Searchlight Helfen Sie dem Simons Searchlight- Team, mehr über die 1q21.1-Deletion zu erfahren, indem Sie an unserer Studie teilnehmen. Hier können Sie mehr über das Projekt erfahren und sich anmelden.
Externe Forschungsgelegenheit: FaceMatch
FaceMatch ist eine Plattform, die Eltern und Ärzten hilft, zu einer internationalen, sicheren Bilddatenbank sowohl von nicht diagnostizierten als auch von diagnostizierten Kindern in der ganzen Welt beizutragen. *Diese Studie steht in keiner Verbindung zu Simons Searchlight. Erfahren Sie mehr über FaceMatch.
Familiengeschichten
Frühere vierteljährliche Berichte
- Stimme der Gemeinschaft Bericht 2021
- 1q21.1 Löschung Quartal 1 Bericht 2021
- 1q21.1 Löschung Quartal 2 Bericht 2021
- 1q21.1 Löschung Quartal 3 Bericht 2021
- 1q21.1 Löschung Quartal 4 Bericht 2021
- 1q21.1 Löschung Quartal 1 Bericht 2022
- 1q21.1 Löschung Quartal 2 Bericht 2022
- 1q21.1 Löschung Quartal 3 Bericht 2022
- 1q21.1 Löschung Quartal 4 2022/Quartal 1 2023 Bericht
- 1q21.1 Löschung Quartal 2 2023 Bericht
- 1q21.1 Löschung Quartal 3 2023 Bericht
- 1q21.1 Löschung Quartal 4 2023/Quartal 1 2024 Bericht
- 1q21.1 Löschung Quartal 2 2024 Bericht
- 1q21.1 Löschung Quartal 3 2024 Bericht
- 1q21.1 Löschung Quartal 4 2024 Bericht – 2024 Surveys Spotlight
- 1q21.1 Deletion Quartal 1 2025 Bericht – Vineland Spotlight
- 1q21.1 Löschung Quartal 2 2025 Bericht
- 1q21.1 Löschung Quartal 3 2025 Bericht
- 1q21.1-Deletion – Bericht für das 4. Quartal 2025/1. Quartal 2026